

Schrodinger Suites(分子模拟软件)
v2015-2 附安装教程- 软件大小:3328 MB
- 更新日期:2020-04-06 15:42
- 软件语言:简体中文
- 软件类别:辅助设计
- 软件授权:免费版
- 软件官网:待审核
- 适用平台:WinXP, Win7, Win8, Win10, WinAll
- 软件厂商:

软件介绍 人气软件 下载地址
Schrodinger Suites 2015是一款非常强大的药物设计与分析工具,该程序包含了从分子建模程序到更复杂的药物设计软件的多种产品,它还具有用于材料研究的现代化套件,它有材料科学,小分子药物发现,生物制剂,发现信息学和PyMOL套件;在程序中,用户可以通过草绘创建化学结,它具有一个称为Maestro的统一图形用户界面,可为所有计算提供服务;该程序提供了对生物制剂,蛋白质和抗体进行建模所需的所有工具;小分子药物发现套件是一个可以加速潜在客户优化和潜在客户发现的套件,使用户可以在整个发现团队中共享数据和协作设计;支持自动化化学提取器(ACE),可以从PDF 2D线图导入化学结构;支持分子动力学,提供轨迹图新型交互计数分析器;新界面可指导用户使用针对拓扑,形状或药效团特征的共识或基于配体的参考,针对同类或多种配体进行高质量,灵活的配体比对;需要的用户可以下载体验
新版功能
量子力学
新的分析基础集LANL2DZ
新的分析基础集系列ANO-VT-XZ
解析6-31G * / **基础集现在支持第4行元素[
可以通过Jaguar面板中的[Keywords]输入框调用六个新的DFT功能TPSS,SCAN,PKZB,SOGGA,SOGGA11,BOP
二维当量H和C原子的平均NMR化学位移
默认情况下,VCD工作流中更健壮,更快的基于PCM的几何优化
默认情况下,所有热化学计算中的准谐波近似均设置为100cm-1
ANI-1神经网络的Beta版本模仿wB97X-D / 6-31G *输出,可用于包含8个“有机”元素的中性分子的单点计算,几何优化和频率计算
新的命令行脚本ring_chain.py生成环链互变异构体
形状筛选
轻松指定多个bin文件以在一项作业中进行筛选
使用gzip格式或新的Pathfinder Reactant文件格式(* .pfx)的压缩试剂库节省大量磁盘空间
将定义自定义反应的rxn格式文件导入PathFinder
在Pathfinder反应/试剂库中添加了三个Claisen反应变体和两个新的试剂类别,醛-酮可烯化和酯类
蛋白质同源性建模
通过将Biopython升级到版本1.74 [2020-1],可以更强大地从NCBI检索BLAST数据。
在基于Prime知识和基于能量的同源性建模中增加了对Fasta格式对齐文件的支持
低温电磁
(可选)为GlideEM [2020-1]的潜在姿态计算QM姿态应变,Phenix OPLS3e改进和Prime MM-GBSA能量。
工作流程和流水线
包括最新版本的KNIME(v4.1)
从KNIME服务器上载工作流程的新节点将通过LiveDesign运行
新的将图像导出到LiveDesign节点
导出到LiveDesign节点可以创建一个额外的分子列
将模型上载到LiveDesign节点的可用性得到了改进
计算诊断失败的作业信息
阅读器节点保留导航历史记录
软件特色
具有KNIME工作流程的SchrödingerSuites 2015-2(Win / Mac / Lnx)
小分子药物发现套件。从QSAR到虚拟筛选再到结合亲和力预测,全面的小分子药物。Discovery Suite包含所有基于片段,配体和基于结构的药物设计所需的所有工具,以进行潜在顾客发现和优化。
生物制剂套装
这个易于使用的新套件是从头开始设计的,是对建模生物制剂,抗体和蛋白质非常重要的所有工具的第一个完整集合。
材料科学套件
这套创新的新套件为基于量子力学的化学系统模拟提供了功能强大的多功能工具,可在特种化学和材料科学中应用对系统进行分析和优化。
Discovery Informatics Suite
用于协作药物设计的下一代平台使包括医学化学家,生物学家建模人员和IT专业人员在内的多学科团队实时共享,查看和管理数据。
PyMOL
PyMOL是一个在开源基础上由用户赞助的分子可视化系统。请通过购买维护和/或支持订阅来支持这种开放,有效且价格合理的软件的开发。
安装步骤
1、用户可以点击本网站提供的下载路径下载得到对应的程序安装包
2、只需要使用解压功能将压缩包打开,双击主程序即可进行安装,弹出程序安装界面
3、可以根据自己的需要点击浏览按钮将应用程序的安装路径进行更改
4、弹出以下界面,用户可以直接使用鼠标点击下一步按钮
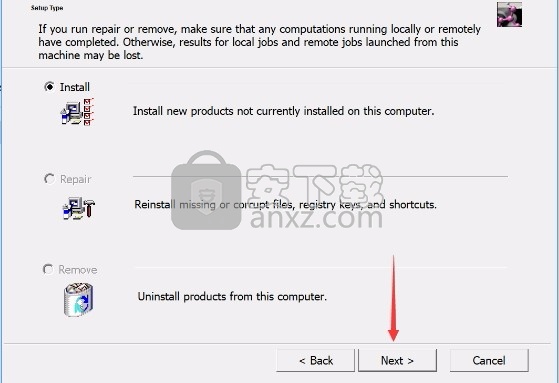
5、弹出以下界面,用户可以直接使用鼠标点击下一步按钮,可以根据您的需要不同的组件进行安装
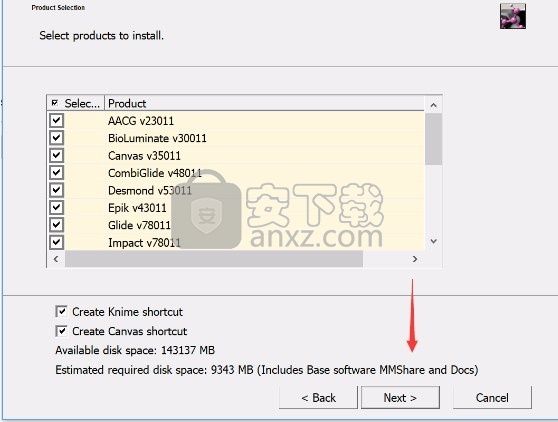
6、现在准备安装主程序,点击安装按钮开始安装
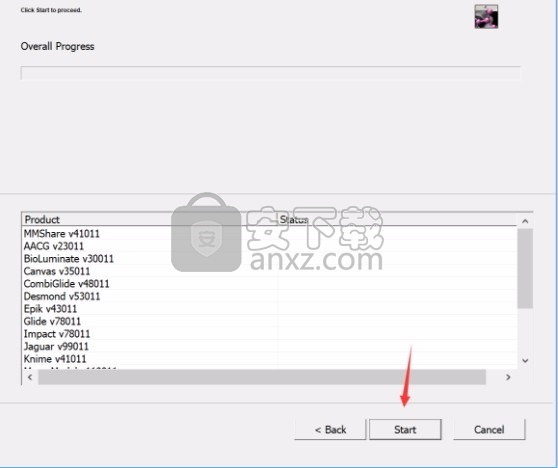
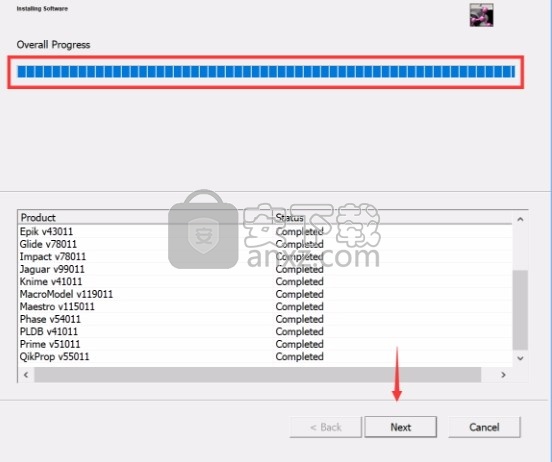
7、弹出应用程序安装进度条加载界面,只需要等待加载完成即可
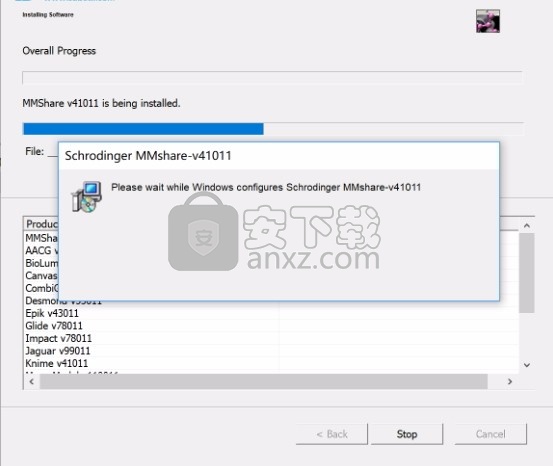
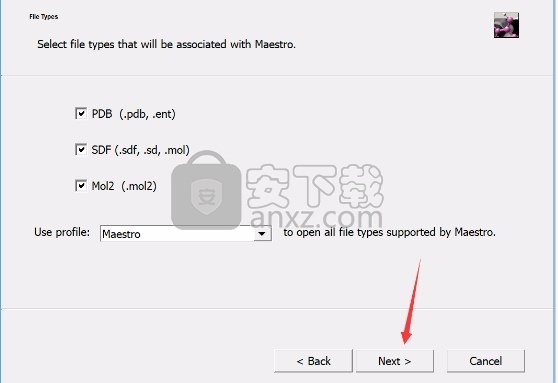
8、程序安装完成后,会弹出文件选择界面,根据需要勾选适配文件类型,点next
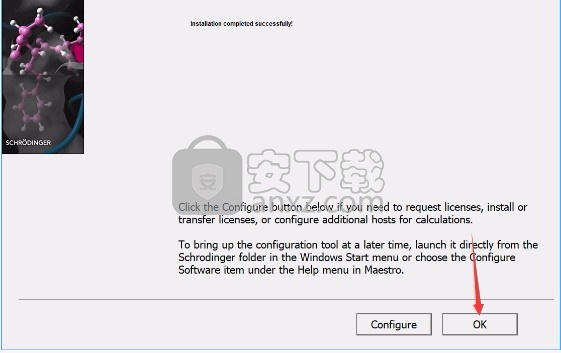
9、根据提示点击安装,弹出程序安装完成界面,点击完成按钮即可
方法
1、程序安装完成后,先不要运行程序,打开安装包,将patcher文件夹内的patcher.exe复制到安装目录(默认:C:\Program Files\Schrodinger2017-4)
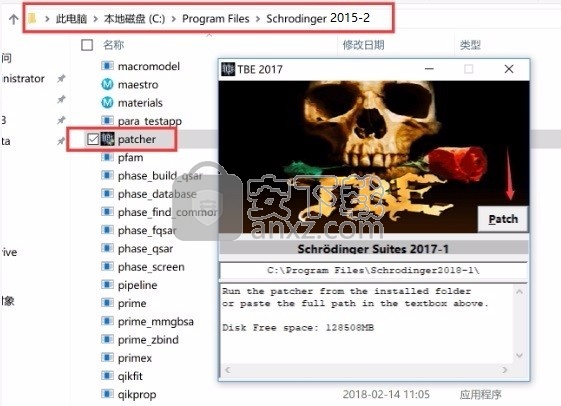
2、然后在程序安装路径下运行注册机,弹出对应的界面后,点patch完成
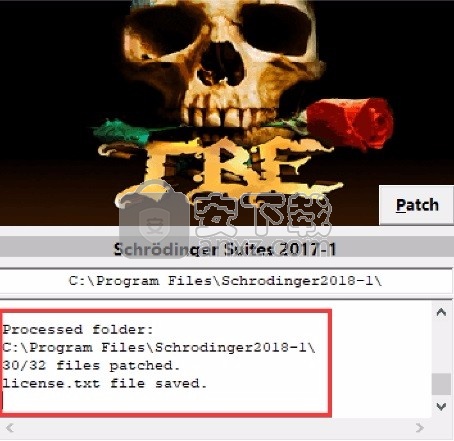
3、点开始菜单,这么多软件都可以免费使用了,双击应用程序将其打开,此时您就可以得到对应程序
使用说明
划分数据集
为了更好地理解五种方法的局限性和优势,我们尝试使用三种方法对数据集进行划分。第一种方法根据突变是保留还是改变蛋白质的净电荷来划分数据集。第二种方法根据突变是否实质上改变了残基占据的体积(大→小或小→大)对数据进行划分。如果通过> =丙氨酸->缬氨酸突变的体积改变侧链的体积,我们将体积变化分类为“显着”。最后,我们使用野生型侧链的相对可及表面积来确定蛋白质表面上发生的突变与掩埋在蛋白质中的突变,以确定其是否暴露于溶剂中[ 23]。
出于几个原因,涉及通常在中性pH(D,E,R,K)下带电的残基的突变可能具有挑战性。例如,取决于蛋白质环境,带电荷的残基可以占据不同的质子化状态,这可以显着改变其物理化学性质。另外,带正电荷的残基,例如赖氨酸和精氨酸,相对于其他氨基酸含有大量的可旋转键,使得预测这些带正电荷的残基的侧链构象具有挑战性。由于面临相同类型的挑战,也已记录了其他基于物理学的算法,难以预测改变蛋白质净电荷的突变的稳定性[ 24]]。因此,我们评估了去除改变蛋白质净电荷的突变后,Prime的性能是否会提高。
正如看到的图4A,总理获得的0.75上的突变改变净电荷的蛋白的AUC。出乎意料的是,这种性能与Prime在整个数据集上的性能(AUC = 0.75)相当,并且与在包含蛋白质净电荷变异的数据集上的性能非常相似(AUC = 0.74,图4B)。只有可访问性度量标准显示出以这种方式划分数据集的显着敏感性,整个保留电荷集的AUC为0.66,而认为改变蛋白质净电荷的突变的AUC为0.80。
我们将数据集划分为从大到小的突变,从大到小的突变,以确定Prime中侧链弛豫方案的增加的灵活性是否会从小到大的突变受益。从小到大的突变被定义为侧链体积比丙氨酸到缬氨酸突变增加更多的突变(相反,从大到小的突变被使用)。有趣的是,Prime中的5分细化方案对从小到大突变的性能产生了负面影响,将Prime的AUC从0.78降低到0.69(图5)。同样,也许并不出乎意料的是,增加的灵活性并没有提高Prime对大到小突变的性能。这表明相对于允许结构的侧链的微小改变,不干扰晶体结构是有益的。凯洛格(Kellogg)等人先前已经观察到了这一点。修改Rosetta能量函数的采样协议时[ 25 ]。还值得注意的是,相对于整个数据集(AUC = 0.71),BLOSUM62替换矩阵在从小到大的突变(AUC = 0.63)方面挣扎。
蛋白质表面的位置通常比蛋白质隐蔽位置的突变更能耐受突变[ 26 ]。因此,我们对本研究中测试的模型的性能是否会对突变发生在蛋白质表面或隐性位置敏感(图6)感兴趣。)。如果野生型氨基酸的溶剂暴露百分比小于或等于5%,则将位置定义为隐性。如果野生型残基的溶剂暴露百分比> 20%,则将位置定义为暴露于溶剂。可以预见的是,可访问性度量不能很好地分类掩埋位置的功能破坏突变,从而使AUC值接近随机值(AUC = 0.47)。相比之下,BLOSUM62矩阵在将突变分类为掩埋位置的功能破坏方面表现出色,导致AUC为0.84。Prime在掩埋集上的性能非常接近其在整个数据集上的性能,导致AUC为0.78,而整个数据集的AUC为0.75。Prime中增加的灵活性并不能帮助Primer掩埋突变,导致AUC为0.73。有趣的是 所有算法在分类蛋白质表面的破坏性突变方面都非常困难。AUC的范围从0.61到0.67。
Prime中的MM-GBSA方法能够胜过本研究中测试的用于预测功能破坏性突变的所有其他方法。而且,也许更重要的是,与本研究中测试的其他方法相比,Prime在数据集的各个部分上的工作更为稳健。Prime广泛的一致性能可能反映了Prime是基于物理的方法这一事实。除表面暴露的突变外,Prime的AUC值范围为0.74至0.80,范围仅为0.06。这等于整个数据集上95%置信区间的范围(图2)。但是,BLOSUM62矩阵的AUC值在0.63至0.84的范围内。这是0.21的变化,该值比Prime的变化大三倍以上。从可访问性度量标准获得的AUC值的范围甚至更大,范围从0.47(几乎是随机模型)到0.81。与Prime相似,FoldX中的计分功能使用基于物理学的术语。因此,与Prime相似,FoldX也实现了一致的性能,其AUC值介于0.67至0.72之间。但是,Prime始终能够在数据集的所有分区上胜过FoldX。
除了对Prime进行基准测试之外,我们还尝试通过在要突变的残基的5分之内的蛋白质侧链中引入灵活性来改善Prime的性能。有趣的是,这会降低数据集所有部分的性能,即使是对于显着增加要变异的侧链数量的突变而言也是如此。其他人在验证蛋白质稳定性评分功能时也有类似的发现。正如在Kellogg等人中观察到的那样,要适当地将蛋白质柔韧性引入Rosetta能量函数,需要使蛋白质的主链原子与侧链运动一起移动[ 25 ]。因此,使用分子动力学未来的研究为基础的方法,例如自由能微扰[ 27,28允许完全蛋白质具有柔性的,也应进行基准测试,以确定这些方法可以基于蛋白质稳定性的变化预测功能性破坏突变的程度。
在讨论Prime在预测将破坏功能的突变的通用性时,讨论其缺点也是至关重要的。素数在表面暴露位置上挣扎,仅获得0.65的AUC(图6)。但是,值得指出的是,当Prime预测表面突变确实会使蛋白质不稳定(从而破坏功能)时,该预测可能是正确的,如图7中使用两个单独的截断值的高精度和特异性得分所示。主要。Prime的局限性在于,当它预测突变不会破坏稳定性(和功能)时,这可能是正确的,也可能是不正确的,这体现在两个不同Prime临界值的低灵敏度值上。
表面突变的主要挑战是这些突变具有多种破坏功能的机制。例如,表面上的半胱氨酸突变可能参与二硫键,从而阻止了蛋白质发挥其功能。这可能就是为什么半胱氨酸突变导致功能破坏的可能性比中性/有益突变高10倍的原因。另外,蛋白质表面的突变会干扰关键的蛋白质-蛋白质相互作用或蛋白质-底物相互作用,而这仅凭蛋白质单体的晶体结构无法预测。但是,在β-内酰胺酶的情况下,我们可以将β-内酰胺酶的晶体结构与酰化过渡态类似物复合使用,以近似估计对天然底物的结合亲和力的变化。为了做到这一点,同时仍然考虑到蛋白质稳定性的变化,我们采用了预测的稳定性变化和预测的每个突变亲和力变化之间的最大Prime评分。这种方法的范例是,如果预测突变的稳定性有微小变化,但预计会显着破坏底物结合亲和力,则该突变仍将被分类为功能破坏,反之亦然。不幸的是,这并没有显着改善结果,将AUC值从0.74更改为0.75(这种方法的范例是,如果预测突变的稳定性有微小变化,但预计会显着破坏底物结合亲和力,则该突变仍将被分类为功能破坏,反之亦然。不幸的是,这并没有显着改善结果,将AUC值从0.74更改为0.75(这种方法的范例是,如果预测突变的稳定性有微小变化,但预计会显着破坏底物结合亲和力,则该突变仍将被分类为功能破坏,反之亦然。不幸的是,这并没有显着改善结果,将AUC值从0.74更改为0.75(图8)。结果,似乎对于该数据集,对底物的亲和力丧失起着名义上的作用。
除了表面突变的挑战外,还有其他因素会阻碍通过蛋白质稳定性预测来预测功能性破坏突变。例如,已经显示了分子伴侣分子指导蛋白质的折叠,其展开状态实际上比折叠状态的能量要低[ 29 ]。这是通过动态捕获处于折叠状态的蛋白质来完成的。令人着迷的是,已知这些分子伴侣对蛋白质的遗传变异体具有异质作用,导致蛋白质稳定性和功能之间的联系进一步模糊[ 30 ]。
应用Prime预测功能破坏突变的另一个重要限制是获得目标蛋白质目标的准确结构的能力。在这项研究中,1BTL晶体结构为建立基于β-内酰胺酶蛋白的结构模型提供了一个有希望的起点,只需要添加和优化氢原子即可。然而,尽管蛋白质数据库很大,但大多数蛋白质尚未结晶[ 31 ]。同源性建模提供了一条途径,可以扩展与已经结晶的蛋白质序列密切相关的蛋白质的蛋白质数据库,未来的研究应探讨Prime如何使用同源性模型预测功能性破坏突变的程度。
最后,使用单层神经网络,我们将整个β内酰胺酶数据集(即可访问性,BLOSUM62和Prime Max)中性能最好的预测变量集成到了单个机器学习(ML)模型中。我们通过随机采样990个单点突变的一半并测试其余一半来训练ML模型。有趣的是,模型获得的AUC为0.84(图8)。尚不清楚ML模型是否会推广到β-内酰胺酶数据集之外。但是,它展示了在单个分数下统一模型的能力。这样的方法可以帮助集成模型,这些模型捕获了其他破坏蛋白质稳定性之外的功能的机制,例如上述方法。最后,尽管有上述限制,但蛋白质稳定性的准确模型确实捕获了β-内酰胺酶数据集中的大量功能破坏性突变。
材料和方法
准备1BTL和1AXB晶体结构
Schrodinger的蛋白质制备向导(PrepWizard)用于制备Prime的所有PDB结构[ 32 ]。PrepWizard分配键顺序,预测质子化状态,采样Asn / Gln / His翻转状态,去除选择的结晶水,优化H键网络并最小化结构。除-propka_pH标志(设置为pH 7.2)外,所有参数均使用默认值。使用了以下命令行:
$ SCHRODINGER / utilities / prepwizard -fillsidechains -propka_pH -NOJOBID
运行残留扫描
缺省情况下,使用了BioLuminate(18)18–3版中的残留扫描功能,为此,通过以下命令行使用了默认采样协议:
$ SCHRODINGER /运行$ SCHRODINGER / mmshare- v32017 / python / scripts / residue_scanning_backend.py作业名$ jobname -fast-res_file -receptor_asl -refine_mut prime_residue -dist 0.00 -NJOBS 1 –NOJOB
由于残留扫描应用程序的限制,打破结构中共价键的突变的Prime评分为1000.0。
拆分β-内酰胺酶数据集
如上所述,已使用三种方法对β-内酰胺酶数据集进行了划分。第一种方法将改变蛋白质净电荷的突变与保留蛋白质净电荷的突变分开。为了产生改变净电荷的突变的数据集,使用了涉及赖氨酸,精氨酸,天冬氨酸和谷氨酸的突变。如果残基突变为这些氨基酸或从这些氨基酸突变,则认为它们会改变蛋白质的净电荷,除非突变保持为正(即赖氨酸变为精氨酸,或精氨酸为赖氨酸)或保持负(即天冬氨酸变为赖氨酸)。谷氨酸或谷氨酸为天冬氨酸)。如果突变不涉及赖氨酸,精氨酸,天冬氨酸或谷氨酸,则认为它们可以保留蛋白质的净电荷。
为了用显着增加(从小到大)或减少(从大到小)突变的数据集分割数据集,我们使用了Darby和Creighton(1993)报道的范德华体积[ 33 ]。大于丙氨酸至缬氨酸突变的体积变化(37 ^ 3)被认为是体积的显着变化,反之亦然。考虑了更严格的过滤器,但是,这些过滤器导致了不可靠的小型数据集。
最后,为了将突变归类为埋藏或暴露于溶剂中,针对野生型结构中的每个位置计算相对溶剂可及表面积。根据Miller等人的方法,通过将溶剂可及性(Accessibility)指标除以三肽中该氨基酸的最大溶剂可及表面积来完成。(1987)[ 23 ]。相对溶剂可及表面积<= 0.05归为埋藏。并且认为> 0.20的相对溶剂可及表面积是溶剂暴露的。该分析排除了甘氨酸残基。
人气软件
-

南方cass 65.9 MB
/简体中文 -

迈迪工具集 211.0 MB
/简体中文 -

origin(函数绘图工具) 88.0 MB
/简体中文 -

OriginLab OriginPro2018中文 493.0 MB
/简体中文 -

探索者TssD2017 417.0 MB
/简体中文 -

mapgis10.3中文(数据收集与管理工具) 168.66 MB
/简体中文 -

刻绘大师绿色版 8.32 MB
/简体中文 -

SigmaPlot 119 MB
/简体中文 -

keyshot6 1024 MB
/简体中文 -

Matlab 2016b 8376 MB
/简体中文
 女娲设计器(GEditor) v3.0.0.1 绿色版
女娲设计器(GEditor) v3.0.0.1 绿色版  iMindQ(思维导图软件) v8.1.2 中文
iMindQ(思维导图软件) v8.1.2 中文  Altair Embed(嵌入式系统开发工具) v2019.01 附带安装教程
Altair Embed(嵌入式系统开发工具) v2019.01 附带安装教程  avizo 2019.1(avizo三维可视化软件) 附安装教程
avizo 2019.1(avizo三维可视化软件) 附安装教程  ChemOffice 2017 附带安装教程
ChemOffice 2017 附带安装教程  绘图助手 v1.0
绘图助手 v1.0 











