

Schrodinger Suites 2017-4 win/linux/mac三版本
附带安装教程- 软件大小:15012 MB
- 更新日期:2020-04-06 15:02
- 软件语言:简体中文
- 软件类别:辅助设计
- 软件授权:免费版
- 软件官网:待审核
- 适用平台:WinXP, Win7, Win8, Win10, WinAll
- 软件厂商:

软件介绍 人气软件 下载地址
Schrodinger 2017-4是一套非常完善的药物分析与药物设计软件包,内置了多个功能模块,并且不同的功能模块代表的功能都不一样,可以为用户提供非常多的服务;Schrцdinger软件套件是一种使用配体和基于结构的方法的药物设计软件,该软件提供准确,可靠和高性能的计算技术,以解决生命科学研究中的现实问题,它为新型候选药物的设计,选择和优化提供了卓越的解决方案和服务;内置的预测模型将使药物发现科学家能够在发现过程的早期评估化合物的特性,并选择具有最佳特征的候选药物;完善的软件预测能力使科学家能够加快他们的研发活动,降低研究成本并做出其他计算或实验方法无法实现的新颖发现;需要的用户可以下载体验
新版功能
Maestro图形界面
从序列[2017-4]构建肽-简化的Pose Viewer面板[2017-4]-导出到电子表格时的控制数据精度[2017-4]-简化的项目历史面板[2017-4]-更好地控制作业名称的自动递增[2017-4]-在项目表中取消选择重复值的选项[2017-4]-轨迹查看器的速度提高了2倍[2017-4]-轨迹查看器现在支持工作区功能区的更改[2017 -4]-从结构层次结构创建通用定制集的选项
使用FEP +面板[2017-4]预测可逆共价结合配体的相对结合亲和力。自动检测共价结合的配体
生成和编辑FEP图
支持膜结合蛋白(例如GPCR)
FEP子作业的吞吐量提高了25-30%[2017-4]-FEP + Panel的多项改进[2017-4]。将实验值注释为测定的顶部/底部
显示Maestro中每个边缘的代表性结构/轨迹对
利用SMARTS模式定义的定制核增强不对称取代环的采样
使用多个CPU内核生成速度图
用户警告Bennett误差超过0.4 kcal / mol
分子动力学
径向分布函数(RDF)计算现在可以通过作业控制提交给远程服务器
Macrocycles
新的Macrocycle Conformer稳定性小组对大环接头进行高通量筛选,以鉴定导致相似结合的已知活性配体[2017- Pharmacophore Modeling-
自动或手动比对药效团假说以及所有相关信息,例如筛选的配体和排除卷[2017-4] 列举
进行生物甾体替代[2017-4]。使用一组233个转换,包括
酸,酯,叔丁基,羰基,酰胺,苯基等
轻松修改或添加为默认生物等位基因,编码为“反应SMARTS”
指定不执行等位基因替代的不可变分子
使用新的Hit Expansion面板[2017-4]执行按目录的模拟搜索。通过虚拟筛选或HTS进行命中的快速跟进
经验性和基于QM的pKa预测
通过识别Jaguar pKa中的官能团使用加权pKas [2017-4] 量子力学
计算非谐振动频率[2017-4]-Compute Huang -Rhys因子[2017-4]-新的QM Conformer和Tautomer Predictor界面
蛋白质制备
改善了蛋白质可靠性报告面板的可用性[2017-4] 工作流程和流水线
更改从打开时通过的KNIME首选项和其他选项艺术大师[2017-4]
软件特色
Schrodinger Suites是领先的计算化学软件。该计划为生命科学和材料科学的所有分支提供完整的解决方案和服务。
公司根据客户的具体要求建立了该软件,帮助研究人员更加接近于改善人体健康和提高生活质量的目标。该产品包括从分子建模到药物设计和制造的一系列工具和程序。
这个应用程序有很多的发现和生产新的材料,研究人员可以使用这个程序来学习和获得这个领域的经验。
您可以构建材料的化学结构,在一个简单而美丽的图形环境中,可以完成计算的类型。
在生物学建模中,我们可以建模各种生物系统,如蛋白质,抗体等。在这组工具中有发现信息套件,
它使用这个工具为所有的项目团队成员与团队的其他成员分享他们的研究成果。
这个程序的重要优点是它的图形性质。
所有建模任务都可以通过图形化和通过草绘和改变不同窗口中的值来完成。这将减少用户错误和舒适度。
安装步骤
1、用户可以点击本网站提供的下载路径下载得到对应的程序安装包
2、只需要使用解压功能将压缩包打开,双击主程序即可进行安装,弹出程序安装界面
3、可以根据自己的需要点击浏览按钮将应用程序的安装路径进行更改
4、弹出以下界面,用户可以直接使用鼠标点击下一步按钮,可以根据您的需要不同的组件进行安装
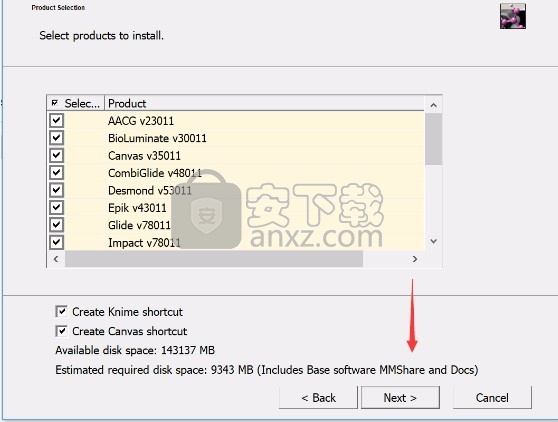
5、弹出以下界面,用户可以直接使用鼠标点击下一步按钮,可以根据您的需要不同的组件进行安装
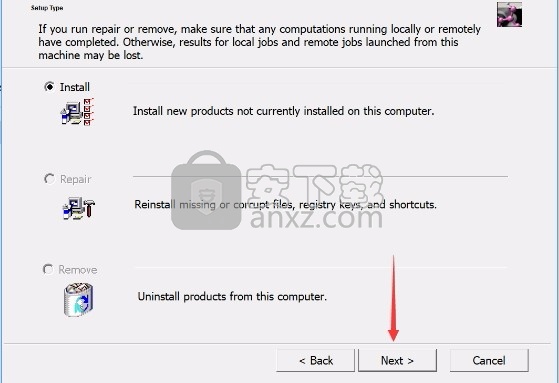
6、现在准备安装主程序,点击安装按钮开始安装
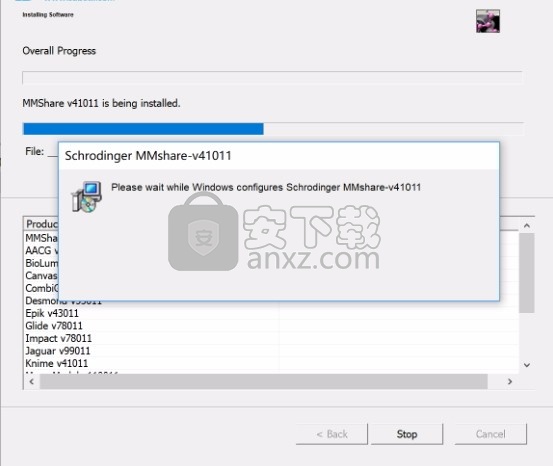
7、弹出应用程序安装进度条加载界面,只需要等待加载完成即可
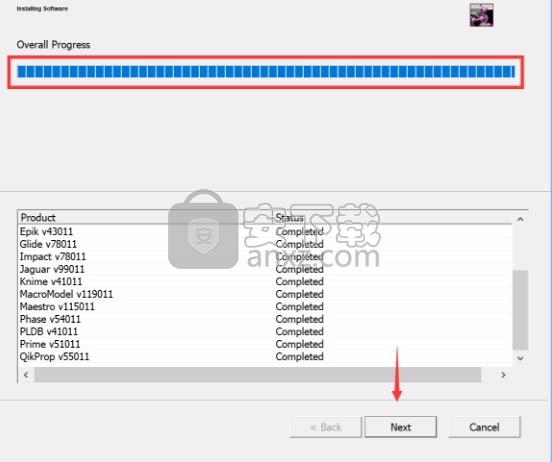
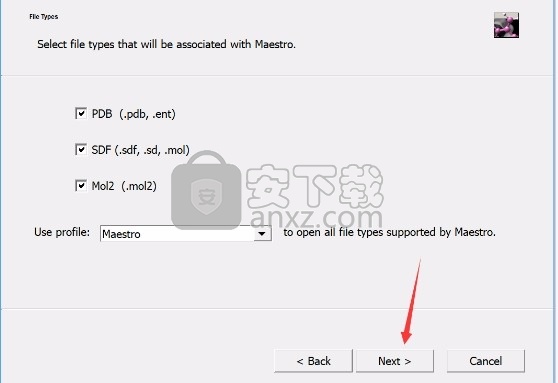
8、程序安装完成后,会弹出文件选择界面,根据需要勾选适配文件类型,点next
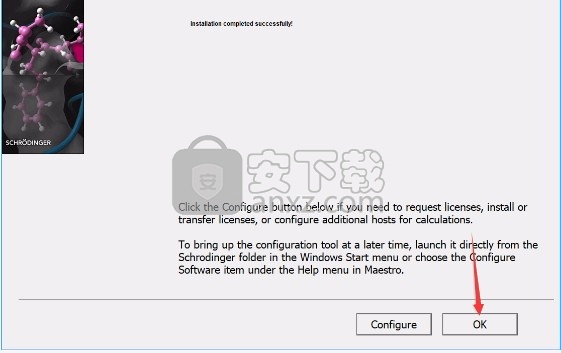
9、根据提示点击安装,弹出程序安装完成界面,点击完成按钮即可
方法
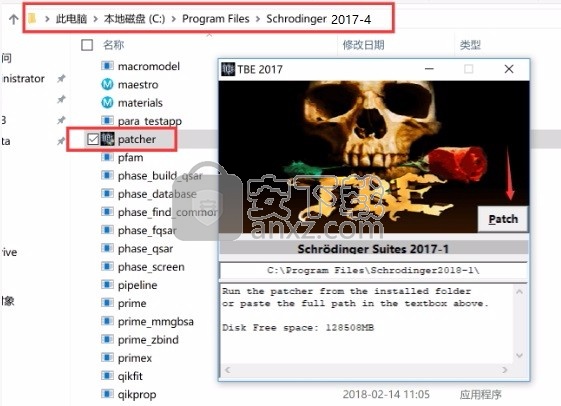
1、程序安装完成后,先不要运行程序,打开安装包,将patcher文件夹内的patcher.exe复制到安装目录(默认:C:\Program Files\Schrodinger2017-4)
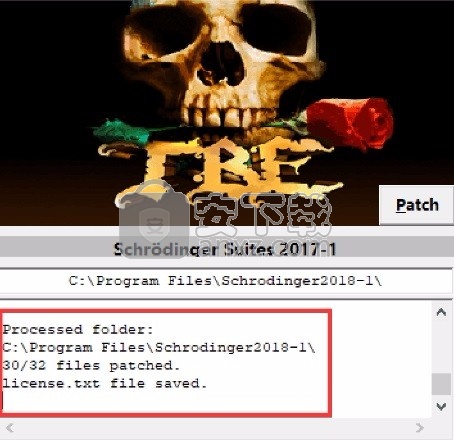
2、然后在程序安装路径下运行注册机,弹出对应的界面后,点patch完成
3、点开始菜单,这么多软件都可以免费使用了
4、完成以上操作步骤后,就可以双击应用程序将其打开,此时您就可以得到对应程序
使用说明
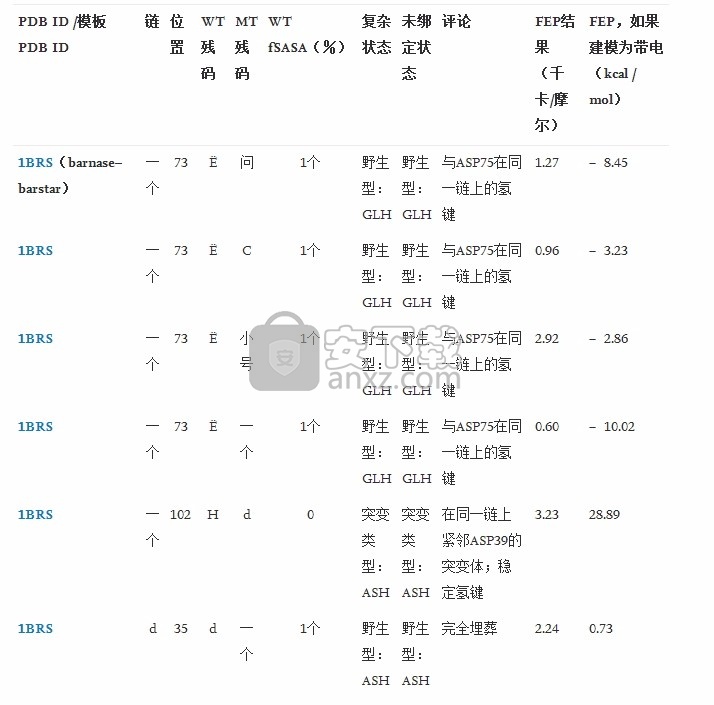
蛋白质-蛋白质界面中具有FEP的电荷改变序列突变的相对结合亲和力预测
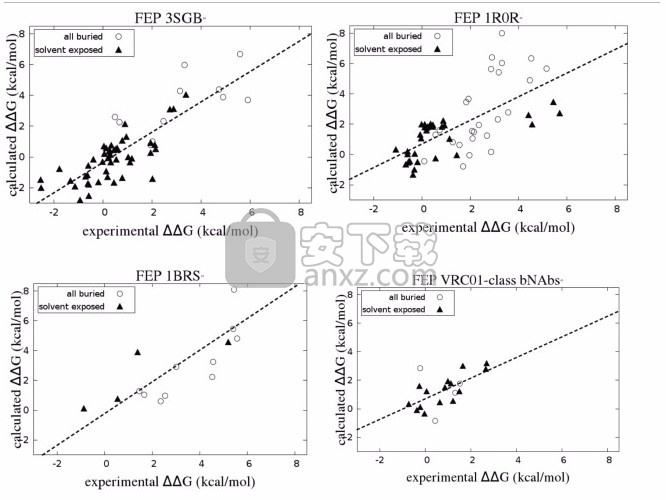
“埋藏”残基突变不包含明显有利于结合的情况,并且除四个以外的所有情况均显着降低结合亲和力(降低超过0.5kcal / mol);自由能量变化的幅度通过FEP确定为0.52来预测。然而,这些情况的RMSE很大,为1.79 kcal / mol。我们认为,这是由于无法在相对较短的FEP轨迹中模拟蛋白质或蛋白质-蛋白质结合模式的显着构象变化。但是,如上所述,在实际项目中并不重要,在该项目中,目的是产生更强的结合蛋白来确定突变是否会引起+的自由能变化 2或+ 5 kcal / mol; 无论哪种正确,均不值得通过实验进行测试。因此,简单地将拟议的突变位点分类为隐性似乎(至少基于该数据集)足以排除该位点的电荷变化突变。我们注意到,在这种情况下,mm-GB / SA在测试案例中的表现非常差,测定系数小于0.1,RMSE非常大(2.8 kcal / mol)。
对于“暴露在溶剂中”的站点,FEP显示的RMSE为1.22 kcal / mol,更为合理,相关系数为0.53。相比其在掩埋残留物上的性能,mm-GB / SA也显示出明显的改善。但是,确定系数的提高几乎完全归因于某种能力,可以将高度不利的突变与数据集的其余部分区分开(比对优先突变或近乎有利的突变进行排序要容易得多的问题)。为了说明这一点,我们在表3中给出了未埋藏数据集的测定系数,其相对实验结合亲和力截断了+ 1大卡/摩尔 对于此数据集,FEP得出的确定系数为0.39,而mm-GB / SA显示接近于零的相关性。这些结果表明,mm-GB / SA(以及可能的其他相关方法)仅在实践中可用作粗滤器,以消除得分很差的突变,而FEP将优先生成高度富集的候选集,包括那些导致重大改进的候选集结合亲和力。
FEP和mm-GB / SA预测与实验结合亲和力变化< 1 kcal / mol的相关性

我们还将结果与使用foldX进行了比较[5],发现总体结果与mm-GB / SA给出的结果非常相似。FoldX的性能类似于mm-GB / SA,对于所有突变,RMSE分别为1.8和1.5 kcal / mol,而只有非埋藏突变,而 mm-GB / SA值分别为1.9和1.5 kcal / mol。此外,在实验值< 1 kcal / mol的情况下,FoldX的测定系数为< 0.01(p = 0.46)。SI中提供了一个图(图S1)。
各种单独系统的结果如图2所示。
FEP建模对完全掩埋位点突变的挑战
考虑到在中性条件下点突变与具有不同优先电荷状态的埋入侧链结合的影响时,如果复合物仍然完全结合,则可能出现以下两种情况之一:(1)复合物的构象发生变化以适应大量溶剂进入界面或(2)侧链的质子化状态发生变化。Barstar上ASP39的突变提供了一个例子,该突变破坏了非常稳定的盐桥网络。尽管复杂的结合比野生型的亲和力低约7 kcal / mol,但该突变体已被结晶(PDB ID 2ZA4)[19] ,其结果显示了蛋白质-蛋白质结合模式的细微变化,从而允许使用水渗透界面并溶剂化在野生型结构中参与盐桥网络的残基。在这种情况下,我们低估了FEP中突变体复合物的结合,但是模拟显然没有收敛,相对结合自由能估计仍在100 ns后趋向于实验值。
在至少某些情况下,复合物实际上可以以相同的构象形成,但是具有相关酸侧链的中性状态。这对于质子化形式的羧酸根侧链(ASP和GLU)特别合理,后者在几何上比基本侧链(ARG和LYS)小,因此更可能在没有空间冲突的情况下适合在被分子占据的空间中使用。野生型构型中的周围蛋白质结构。实际上,测试仪包含许多掩埋的野生型酸残基[请参见支持信息(SI)中的汇总结果表]。在这种情况下,我们会在FEP中对两种状态进行显式建模,在认为侧链在未结合状态下去质子化并在结合时质子化的情况下,并入近似状态惩罚。隐含的结合自由能较低的状态与实验进行比较。如参考文献中所述。[16],AP ķ 一个校正必须的情况下的状态下,未结合的和结合的状态之间改变施加。由于准确地计算P中的极限挑战ķ 一个由埋地残留经历转变,由于当地蛋白质的环境中,我们通过假设在p近似校正ķ 一个转变是非常大的,处于绑定状态的人口完全是质子化的。表2在数据集中包含所有这些指标的值;在所有情况下,RMSE和确定系数都在包括这些情况的全套不确定性范围内,因此将它们包括在内不会影响我们的总体结论。认为突变的酸侧链被质子化的所有情况都非常不利于结合,但是如果天真地模拟为带电,则认为具有野生型酸侧链被质子化的一些情况将导致结合亲和力提高的假阳性预测。 从而使这种对掩埋残留物的分析对于优化性能至关重要。表4列出了所有使用非标准质子化状态的情况。
GLH和ASH分别是指谷氨酸和天冬氨酸中羧酸盐的质子化形式,而GLU和ASP是指它们的去质子化(带电)形式。
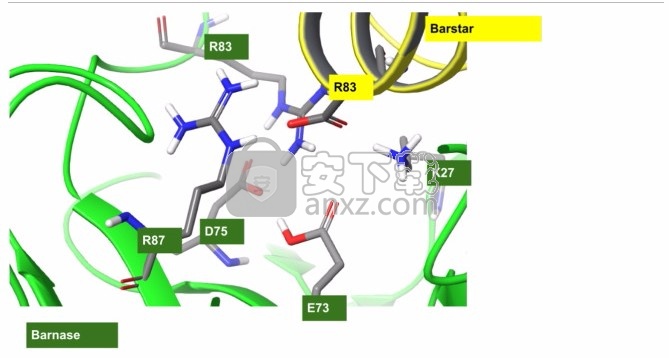
质子化形式的掩埋的酸残基可能特别受人欢迎的一种情况是它非常靠近另一个酸侧链,并且氢键网络涉及质子化的羧酸盐形式与相邻基团。关于这种情况的一个经过充分研究的典型例子是天冬氨酸蛋白酶,特别是HIV 蛋白酶,一种蛋白质同型二聚体,据信其中两个近端催化天冬氨酸之一被质子化了 [30], [31], [32], [33] 。我们认为在此数据集中出现的这种效应的突出例子是野生型Barnase上第 73位的谷氨酸,在FEP轨迹中发现质子化形式,与相邻的羧酸盐侧链(ASP-75或ASP-39,见图 4 )形成稳定的氢键。先前使用双突变周期的实验工作发现此残基与ASP39之间存在强的隐含耦合,他们将其描述为“…令人惊讶,因为Glu73不直接与barstar相互作用,并且在barnase上Glu73与带负电荷的结合表面之间存在静电排斥酒吧之星” [24]。在晶体结构模型1BRS中找不到此配置,但可以在MD中可靠地形成并保持这种配置。使用GLU侧链的带电形式和中性形式并扰动相同的中性侧链进行突变的FEP计算表明,显着因素优选中性形式。从带电形式开始, 对于所有情况,FEP预测与野生型复合物相比,突变体的结合亲和力增加> 2 kcal / mol。从中性形式开始,FEP恢复所有这些突变体的正确分类(不利)。人们还认为,也对barnase起作用的Asp75对该效应负有部分责任,Glu73也被建模为未结合形式的中性,并且在结合时不会引起状态罚分。
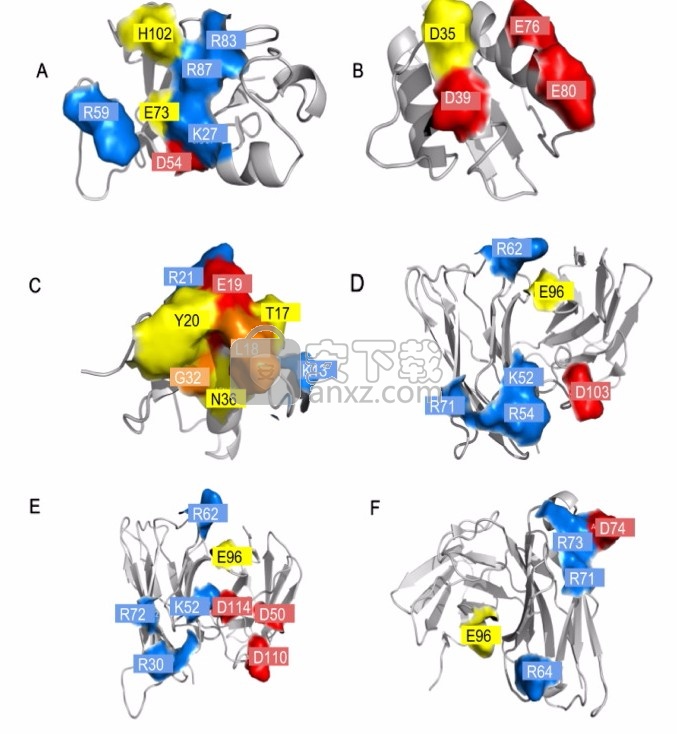
图4。观察到质子化的GLU-73侧链(底部)与去质子化的ASP-75侧链形成稳定的氢键,以稳定barnase和barstar 的复合物中的关键掩埋盐桥网络。
尽管这些特定情况与生物亲和力优化不太可能相关,但是使用FEP模拟评估可电离残基的质子化状态(如上所述)可能具有潜在的应用。蛋白质界面掩埋部分中质子化状态的分配是一个非常困难的问题,该方法可以与少量实验诱变数据结合使用,以确定可能的首选质子化状态分配。
模型与方法
电荷扰动的FEP协议
由于多种原因,很难准确计算出改变残基净电荷的突变的结合自由能的变化。首先,大多数MD模拟采用有限尺寸的模拟盒来采用周期性边界条件来近似宏观系统的行为,并且周期性边界条件引入了人工产物,用于带电溶质的静电势能计算。伪像可以分解为溶质及其周期性图像之间的净电荷相互作用,由于模拟盒的有限大小而导致的溶质欠溶剂化以及在不同系统的模拟中零静电势的不一致性 [14]。其次,通常在含盐的缓冲溶液中进行实验性结合亲和力测定,带电溶质之间的结合亲和力主要取决于缓冲溶液的离子强度,很难准确建模。第三,改变残基净电荷的突变通常与其蛋白质结合伴侣和周围的水分子具有非常不同的相互作用,并且当这些差异相互作用导致实质性构象变化时,很难聚合自由能计算。
我们测试了Refs中首次引入的共化学粒子方法的一种变体。[15],[16],并在参考文献中广泛研究了小分子。[16]计算电荷改变残基突变对蛋白质-蛋白质结合自由能的影响。所使用的协议与参考文献中所述相同。[11],将FEP +与OPLS3力场一起使用,除了以下修改以适应共炼金法:
(1)通过引入共炼金颗粒来节省电荷。特别是,对于从中性残基到带电残基的突变,粒子与残基突变共同突变,从中性粒子变为钠离子(突变的残基具有 -1电荷)或氯离子(突变的残基具有+ 1个费用)。从放置在盒子中的离子中随机选择离子,然后突变为中性粒子。离子上没有任何其他限制以防止其进入蛋白质。这样,两个物理最终状态之间的净电荷变化为零,并且消除了静电势能计算中与仿真盒大小相关的伪像,从而将其限制在自由能计算的不确定性范围内[20],我们基于对不同随机初始速度条件下相同扰动的重复试验得出的估计值约为0.5 kcal / mol [11]。
(2)为了模拟缓冲溶液中离子强度对蛋白质与蛋白质结合自由能的影响,在模拟中明确添加了浓度与测定条件的离子强度匹配的钠和氯离子。通过随机替换产生的初始溶剂缓冲液中的水来放置这些离子。
(3)为了增强自由能计算的收敛性,FEP中的λ窗口数 +模拟从电荷保留突变的12个增加到电荷变化残基突变的24个,进一步优化了能量的静电项权重,以最大化相邻Lambda窗口之间的配置空间重叠(请参见SI部分Lambda权重)对于此非均匀的重量表)。此外,在模拟盒中使用了8.5Å而不是5Å的较大溶剂缓冲液。在一起发现,在许多情况下,这些修改可以在相邻的lambda窗口之间始终产生良好的能量重叠,并且随着增加的仿真时间而具有良好的收敛性。此外,使用野生型多部分盐桥中的残基进行扰动的模拟时间要长得多(每个窗口100 ns),聚糖片段保留在模拟系统中[11]。
通过这些技术改进,有效解决了防止对电荷充电残基突变进行精确建模的难题。
电荷变化突变测试案例对FEP的适用性评估
本文使用的两个数据集是系统扫描所考虑的结合界面中每个位置上所有可能的氨基酸残基的示例。在这种方法中,尝试进行突变的子集将涉及将侧链置于可能需要进行重大结构重排才能使蛋白质结合的位置。此类情况通常会严重破坏复合物的结合,因此不可能找到用于炼金术FEP模拟的合理起始几何形状。此外,在这种情况下,FEP不可能能够进行足够的采样以预测结合亲和力的正确变化。
为了确定不可能进行合理的侧链放置的情况,我们对野生型结构中的突变残基和任何可能发生碰撞的相邻侧链进行了隐式溶剂侧链预测。使用OPLS3力场计算预测的侧链构象的内部能量,并将其与来自旋转异构体库的优化旋转异构体的相同能量进行比较。如果系统中产生的侧链内应变能(偏离最佳旋转异构体的能量)超过了野生型系统的5 kcal / mol,则可以得出结论:侧链的合理起始几何构型为不可能和点突变没有被提前进行FEP。选择该阈值作为固定值,该固定值是此处考虑的系统的总结合亲和力的很大一部分(在 -10至 -20 kcal / mol 的范围内)。应该使用的确切阈值可能需要进一步优化。在最初考虑的最初的161例中,这仅影响了11例。SI中的汇总结果表中记录了具体情况。
涉及掩埋残基的突变的分类
对于其余情况,使用fSASA将输入结构中的突变位点分类为掩埋(fSASA < 10%;总共44例)和暴露于溶剂中(fSASA≥10 %; 106例)。fSASA的定义是界面中的残留物经历的三肽构型中最大可能的溶剂可及表面积的分数 [13]。 。我们在这里注意到,所有对结合具有显着有利影响的突变都包含在后一组中,并且在非埋入位置组中,FEP性能良好(RMSE 1.23)。因此,出于以增加亲和力为目标的实际优化项目的目的,我们建议将fSASA用作第二个易于自动化的过滤器,以确定哪些潜在的电荷变化突变可发送至FEP预测。为了科学利益,我们还在这里检查了44个埋藏的案例,并试图了解它们在FEP中的行为。
mm-GB / SA协议
对于这项工作,我们使用了BioLuminate中实施的mm-GB / SA方法(3.0.011版,Schrödinger,LLC,纽约,纽约,2018年)来预测氨基酸突变后蛋白质-蛋白质界面的结合亲和力的相对变化。[34] 。该方法将OPLS力场[35]与隐式溶剂模型VSGB [36] 集成在一起,并且旋转子库和Prime的搜索方法[37]。。构象采样仅限于侧链;蛋白质主链保持固定,并且在此方案中不使用MD采样。可提供所有复合物的输入结构和所有FEP结果的电子表格作为补充,以便读者可以与采用额外采样的mm-GB / SA方法进行比较。OPLS与VSGB模型的集成将键合和非键合项的OPLS建模与VSGB模型的溶剂化和去溶剂化能结合在一起,其中还包含基于物理学的校正项。应该注意的是,没有术语可以改变侧链构型熵,但是尽管有此限制,该方法的执行效果却与其他mm-GB / SA实现一样好 [38]。。对于本研究中探索的系统,mm-GB / SA输入结构中的结晶水被删除,因为它们会稍微降低性能(RMSE从1.95增加到2.09 kcal / mol,r 2从0.22 减小到0.13)。包括结晶水在内的结果可以在表S3中的SI中找到。
Beard 等人先前的工作。[34] 观察到,将原始能量转换为实验能量的最佳斜率取决于系统。因此,按以下方式确定重新定标的Prime能量,以从Prime获得最佳性能。数据集分为四个子集。这些子集中的三个对应于从SKEMPI数据库获得的实验数据的三个子集(即Barnase:Barstar,Subtilisin Carlsberg:OMTKY3和S. griseus 蛋白酶 B:OMTKY3)。数据的最后一个子集对应于所有涉及VRC抗体的实验测量。将VRC抗体数据汇总到一个子集中,以创建具有合理大小和能量分布(19个突变)的数据子集。然后针对Prime预测的相对结合亲和力变化与相对结合亲和力的实验测量值之间的每个数据子集计算最佳拟合线。最佳拟合斜率范围为0.07至0.14。这些线的斜率用于重新调整每个子集的Prime预测能量。然后将这些重新换算的能量用于计算该工作中报告的最终RMSE和r 2值。
盐桥网络采样
FEP在蛋白质-蛋白质界面上的电荷变化突变中相对频繁发生的采样挑战是电荷的变化可能会破坏跨越多个残基的盐桥网络。因此,当对鉴定为参与多个盐桥(包括双齿基序)的残基进行突变时,由于所涉及的强静电力,有望从初始构象中采样到较大的能量壁垒,因此模拟时间延长至100 ns。对于艾滋病在使用同源性模型的bNAb案例中,在对带电残基进行建模的表面上取代中性侧链的位置存在其他不确定性,我们将所有模拟时间延长至100 ns,以实现更完整的采样。此外,如果观察到在模拟轨迹中形成新的盐桥,则使用REST区域中的突变型盐桥伴侣侧链重新进行模拟,以允许从最初形成的盐桥中取样。
此处显示的FEP计算相对便宜,考虑到GPU时间的价格点约为每GPU小时0.40美元,每次模拟的成本约为10-15美元,并且大多数可以在8小时内完成。因此,进行数千或数万次计算以探索对诸如抗体或疫苗之类的生物疗法的潜在改进是可行的。考虑到噬菌体展示等方法的可用性,这种计算会对项目产生什么样的影响 实验性地组合探索序列空间,还有待观察。靶向特定目的突变并至少估计对其他特性的影响的能力对于在实际应用中产生重大影响至关重要,而这些特性必须进行优化才能创建开发候选物。
蛋白质残基FEP的第二种应用方法是深入了解重要的生物识别事件(例如钙黏着蛋白结合)的选择性。钙黏着蛋白的微小序列变化对于识别彼此的不同类型的细胞(并拒绝其他细胞类型)至关重要。原则上,给定足够好的起始结构,FEP可以为这种识别提供准确的原子能级基础。尚未尝试使用FEP在这些方面进行大规模探索。
结合最近获得的小分子配体结合的结果,目前的工作提供了有力的证据,证明了共炼金水方法是将FEP应用于涉及净电荷变化的转化问题的令人满意的解决方案。电荷变化突变的RMSE甚至比中性突变的RMSE更大,即使对于溶剂暴露的位点也是如此。我们怀疑这是由于非平凡蛋白质重组的可能性更大,即使该位点不属于我们在此定义的“埋藏”类别。随着时间的推移,改进的采样协议和简单的蛮力增加仿真长度应解决这些问题。
人气软件
-

南方cass 65.9 MB
/简体中文 -

迈迪工具集 211.0 MB
/简体中文 -

origin(函数绘图工具) 88.0 MB
/简体中文 -

OriginLab OriginPro2018中文 493.0 MB
/简体中文 -

探索者TssD2017 417.0 MB
/简体中文 -

mapgis10.3中文(数据收集与管理工具) 168.66 MB
/简体中文 -

刻绘大师绿色版 8.32 MB
/简体中文 -

SigmaPlot 119 MB
/简体中文 -

keyshot6 1024 MB
/简体中文 -

Matlab 2016b 8376 MB
/简体中文
 女娲设计器(GEditor) v3.0.0.1 绿色版
女娲设计器(GEditor) v3.0.0.1 绿色版  iMindQ(思维导图软件) v8.1.2 中文
iMindQ(思维导图软件) v8.1.2 中文  Altair Embed(嵌入式系统开发工具) v2019.01 附带安装教程
Altair Embed(嵌入式系统开发工具) v2019.01 附带安装教程  avizo 2019.1(avizo三维可视化软件) 附安装教程
avizo 2019.1(avizo三维可视化软件) 附安装教程  ChemOffice 2017 附带安装教程
ChemOffice 2017 附带安装教程  绘图助手 v1.0
绘图助手 v1.0 











