
Sequencher(DNA序列分析工具)
v5.4.5- 软件大小:235 MB
- 更新日期:2020-08-15 11:34
- 软件语言:英文
- 软件类别:信息管理
- 软件授权:修改版
- 软件官网:待审核
- 适用平台:WinXP, Win7, Win8, Win10, WinAll
- 软件厂商:

软件介绍 人气软件 下载地址
Sequencher是国外开发的一款DNA序列分析软件,用户可以在软件新建一个分析计划,也可以在软件使用官方提供的实验模板创建分析工程,模板是一种特殊类型的项目,可以通过选择参数和首选项进行设置,模板可以包含普通序列或参考序列,首选项可以包括功能设置,酶设置和显示设置,使用模板您还可以设置“装配参数”,“按名称装配”手柄和“修剪”参数创建方差表,可以设置同一重叠群中序列的方差表,当您准备开始创建方差表时,可以从重叠群中选择单个序列,或者转到“项目窗口”并单击其图标来选择重叠群,这将选择所有重叠群的序列,Sequencher可以在软件使用自动定序器中的色谱图来帮助您编辑重叠群,支持显示痕迹和二级峰以发现杂合子,支持编辑痕迹的碱基或还原为实验数据,支持显示限制图,您可以通过在“项目”窗口中选择其序列图标并打开编辑器来查看序列或重叠群的限制性图谱。
软件功能
1、SNP 检测
使用 Sequencher 来进行比较对准以便鉴别和报告 SNP 以及突变。例如,调用第二峰..功能分析你的所有序列以寻找潜在的杂合子。你可以控制定义一个杂合子的严格度。你可以从一个杂合子跳到下一个,而仅仅需要在 Base View 里面按一下空格键。你还可以观察一致及其参考序列的转换。一致及其参考序列的区别会列在一个可导出的表格内。参考序列确保从一个装配到下一个装配时,SNP 的数目保持一致。
2、序列装配
Sequencher 的装配算法可以将你的 DNA 片断快速而又准确的装配起来。自觉的工具允许你在数秒内就可以设置好参数并且调整它们。你完全可以不顾方向的装配序列。Sequencher 会比较前端和反转补体方向来装配最可能的 Contig。
3、序列编辑
Sequencher 能够给你足够的信心认为一个序列是绝对正确的。你可以只观察一个序列的色谱图,你也可以从前端或反方向同时查看多个对齐的色谱图。 在你的已对齐数据中滚动是很容易的。你可以使用 Sequencher 的选择工具来高亮不一致或者低质量的区域。
4、自动分析
Sequencher 可对你的数据进行批处理,这个过程是透明、用户可定义、可恢复的,并且 Sequencher 从不为了自动化的目的而破坏你科学结论的正确性。
当你工作在多个序列时,Sequencher 可以:
调用第二峰
调整矢量
调整低质量的尾端
创建一致序列
恢复到实验数据
Sequencher 总是管理两份数据,一份编辑过的,一份则是原始的导入数据。当你应用“Revert to Experimental Data”命令到在你项目中已选定的序列或者在一序列中的选中部分时,你可以撤销所有或者部分编辑动作。
自从 4.5 以后,Sequencher 就包含了一个新的自动化工具,“Assemble by Name”。依靠“Assemble by Name”,你可以选择片段名的一部分作为一个共享的标识符,或者说是“装配句柄”。Sequencher 就可以将选择和名字自动转换为 Contig。例如,仅仅通过一个按钮的点击,你就可以将 90 个文件,45对前端和反向序列转换成 45 个根据你的病人编号命名的 Contig。装配参数的每个变化都会重组你的片段,因此你可以根据克隆编号、日期、引物,或者其他任何你记录在序列名称中的特征,来装配 Contig。
5、矢量调整
自动排序器以基础调用错误生成结果。从 DNA 库克隆的序列通常包括矢量序列、polyA 尾,或其他不相关序列。内含子和引物序列通常会和放大的外显子序列侧面相连。不调整的话,这些污染物会扭曲你的装配和下游分析。 Sequencher 允许你调整低质量或者含糊的数据。“Trim Ends”将令人误解的数据从序列片段的尾部去除。“Trim Vector”将污染序列尾部的特定序列数据移除。“Trim to Reference”剔除超出装配参考序列的序列尾部。
软件特色
关于项目窗口
该项目的概念对Sequencher至关重要。 用户在项目框架内工作。 一个Sequencher项目包括DNA序列和重叠序列(重叠序列的连续比对)的集合,这些重叠序列由这些序列构建而成。 一个项目可以任意大小。
重叠群编辑
DNA测序是一个容易出错的过程,因此来自自动DNA测序仪的碱基检出可能无法正确代表正在测序的样品。这意味着使用自动DNA测序数据时,通常需要检查和编辑重叠群。
Sequencher在用户和数据之间提供了强大的界面,因此您可以根据自己的规范分析和编辑序列和重叠群。您可以直接从一个歧义或分歧转移到下一个歧义或分歧,以分析和覆盖碱基检出错误,从而消除差异和歧义。 Sequencher允许您从许多共识计算中进行选择,并在编辑序列时不断重新计算重叠群的共识。
编辑重叠群
我们解释如何找到需要注意的歧义和低置信度等基础。我们讨论了如何执行编辑,移动和删除碱基和序列,插入缺口或碱基以及根据共识创建新序列。 您将学习如何比较序列以突出显示差异以及如何创建自定义报告。
寻找需要注意的基础
您的数据显示中可能会看到许多问题。 有些将需要检查,也许需要编辑。 这些问题可能表示为N,意见分歧,差距或置信度低。 您可以使用重叠群的共识序列中“选择”菜单中的“下一步”命令来查找任何这些问题区域。
一旦使用了任何Next命令,Sequencher将激活空格键以执行该操作。 如果您连续两次使用菜单命令或快捷键组合,Sequencher会提醒您此更简单的替代方法可用。
方差表
方差表以最简单的形式比较并显示两个序列之间的差异。 差异表中的数据被动态链接到基础重叠群中的数据。
您可以将重叠群中的某些或所有序列与该重叠群的共识序列,参考序列或最上层序列进行比较。 这将生成一个方差表,该表将显示示例序列与您选择的序列之间的差异。 结果可能仅来自一个重叠群,但可以代表数百乃至数千个序列的集合。 如果要执行从头测序,克隆检查或重新测序之类的活动,则可能要使用此形式的方差表。
翻译方差表
如果您有兴趣关注氨基酸水平上的序列或重叠群之间的差异,则应使用翻译变异表。该表与变异表的不同之处在于,它同时显示了密码子及其相关的氨基酸残基。翻译差异表中的每一行总结了相对于示例在给定氨基酸位置上所有选定序列翻译之间的差异。
您还可以创建翻译后的差异表,该表汇总了项目中所选重叠群共有序列的翻译与通用参考序列(翻译的一致性差异表)的翻译之间的差异。当您处理来自多个来源的多个样本,并且已将“按名称组装”与参考序列一起使用时,应使用此形式的“翻译差异表”。
从序列编辑器查看色谱图
Sequencher的色谱图显示与其他编辑器紧密集成。您可以通过打开序列的序列编辑器来查看序列片段的整个原始轨迹。单击按钮栏中的“显示色谱图”按钮,或转到“窗口”菜单,然后选择“色谱图”。您可以垂直(默认)或水平滚动。要更改方向,请单击片段色谱图窗口左下角的相应按钮。
Sequencher contig编辑器使您可以同时查看与共有碱基调用有关的所有色谱图数据。如果您位于重叠群的5'末端,则Sequencher会选择共识中最5'的碱基,并显示所有相关的迹线。如果您在重叠群中的其他位置,Sequencher会在重叠群显示中选择最中心的共识基数,并显示关联的迹线。这使您可以结合其碱基检出来检查任何峰的信号强度,并相应地编辑数据。
安装方法
1、打开Sequencher 5.4.5.exe软件直接安装,这里显示很多PDF文件,自己阅读吧
2、提示软件的安装引导界面,点击下一步
3、软件的安装协议内容,点击接受协议
4、提示软件准备安装界面,点击install执行安装
5、软件的安装进度界面,等待Sequencher 5.4.5.exe安装结束吧
6、Sequencher已经成功安装,点击finish结束安装
方法
1、打开AMPED文件夹,打开Sequencher-RLMServer。将许可服务SequencherServer.exe安装到电脑
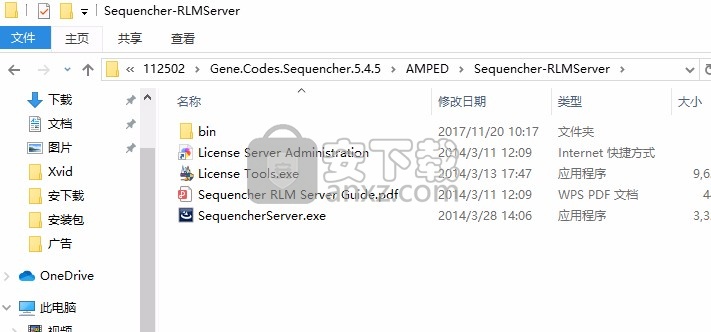
2、提示SequencherServer已经安装结束
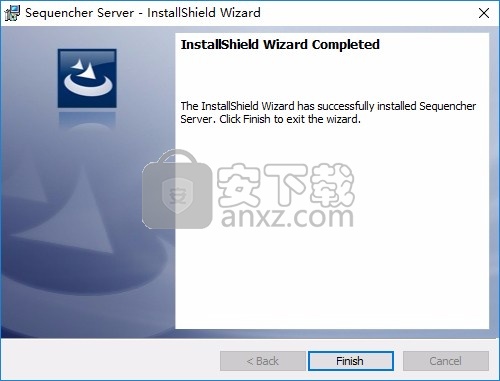
3、进入许可服务安装的地址C:\Program Files (x86)\Gene Codes\Sequencher Server,管理员身份启动Stop RLM Server.bat停止当前的服务
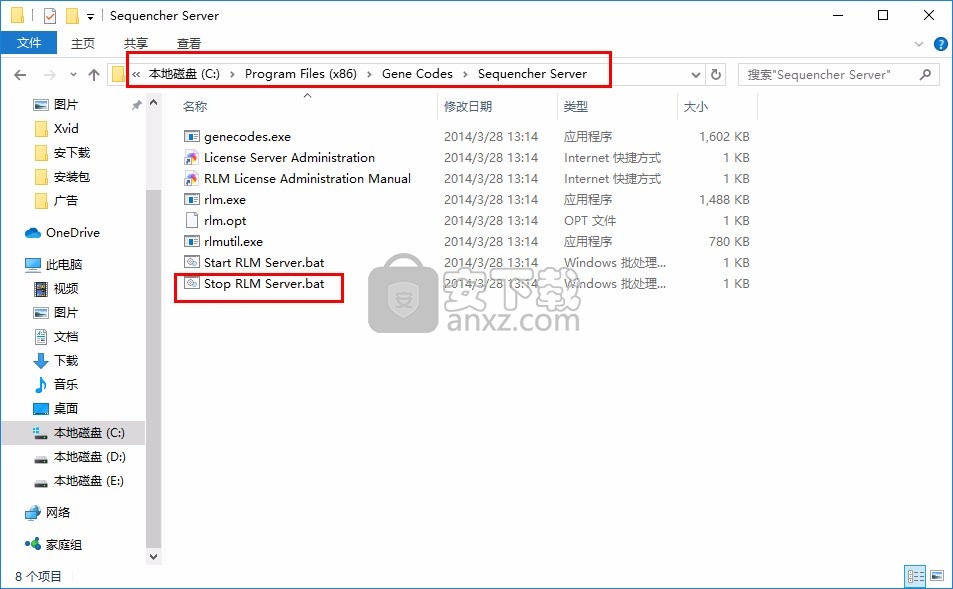
4、将rlm.exe,genecodes.exe和genecodes.lic从AMPED目录复制到Sequencher Server目录,点击替换
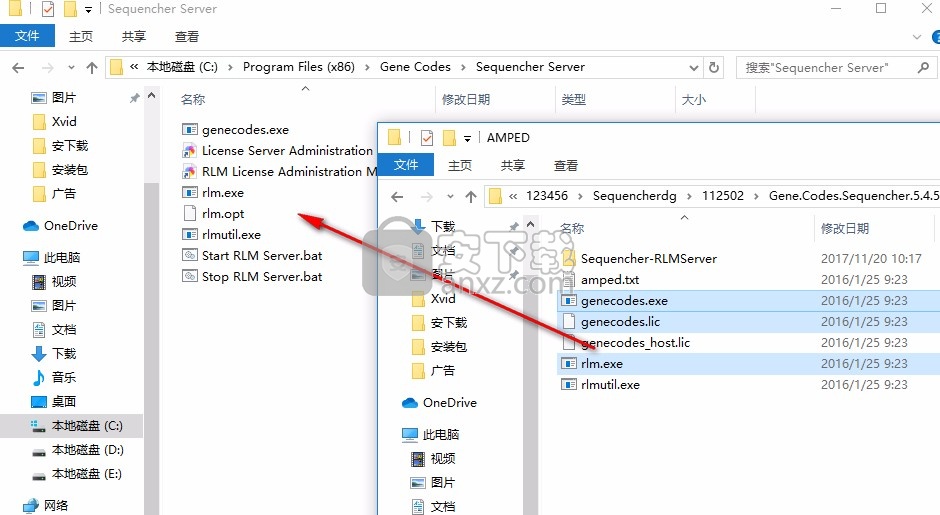
5、管理员身份点击Start RLM Server.bat启动许可服务
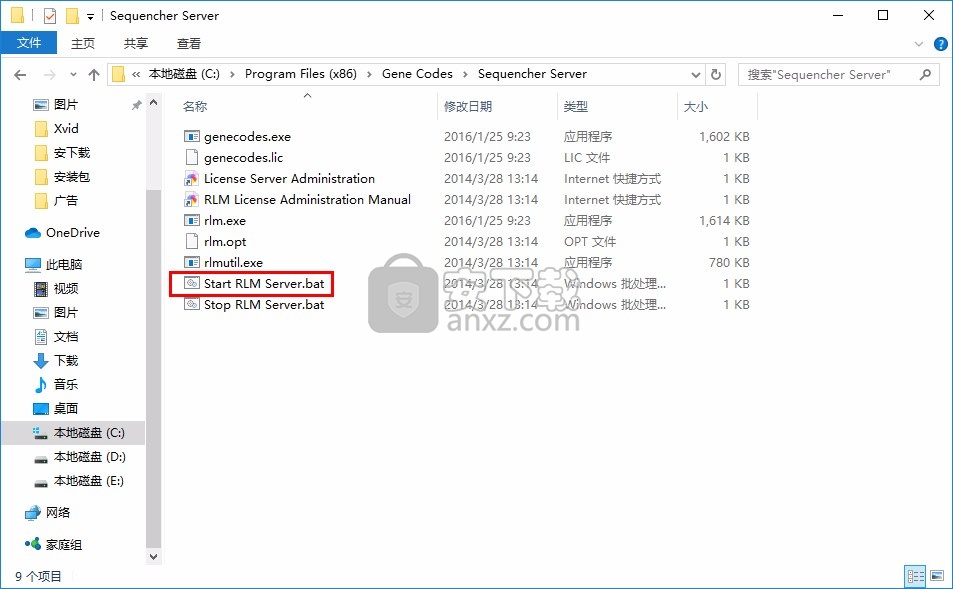
6、打开安装结束的Sequencher软件,点击 Activate Sequencher激活软件
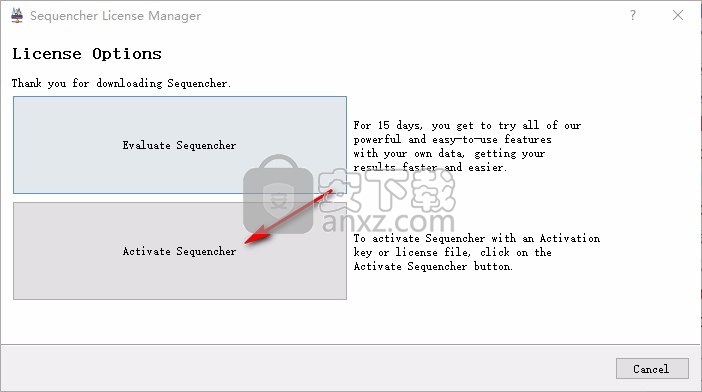
7、点击install安装许可证功能
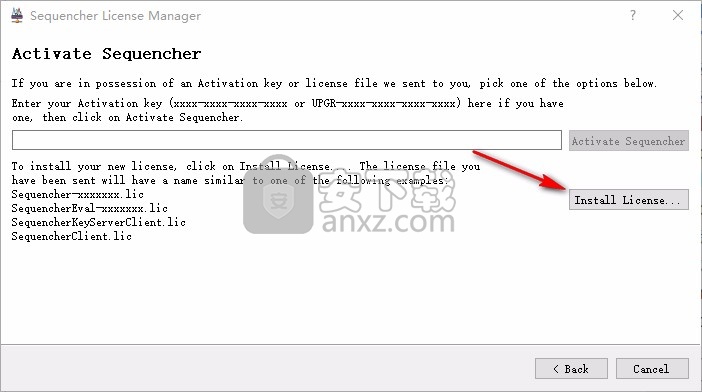
8、打开genecodes.lic,点击install就可以安装
9、安装许可证结果,提示Thank you for purchasing Sequencher,感谢您购买Sequencher
10、点击右下角的start按钮就可以启动主程序
11、软件是英文,如果你会使用Sequencher就开始工作吧,可以查看Sequencher的帮助内容
使用说明
大会策略
Sequencher为您提供了执行序列组装的强大选择。 在许多情况下,默认选项会表现良好。 但是,实验差异会影响您的数据,因此有时您可能需要更改装配参数。 以下是组装数据的基本策略。
•从严格的参数开始,以最小化不正确匹配的可能性。 选择要组装的序列,然后使用“自动组装”选项。 自动组装依赖于详尽的搜索和比较算法。
•如果某些序列未汇编,请降低参数的严格性,然后再次尝试自动组装。
•如果所选参数变得足够宽松,以至于“自动装配”过程可能导致科学上无法支持的路线,则应切换到交互式路线,以便您可以更好地控制过程
设置组装条件
设置装配参数
要更改参数设置,请选择项目窗口。 然后在“项目”窗口中单击“装配参数”按钮,或转到“重叠群”菜单并选择“装配参数”命令。 Sequencher将显示如下图所示的对话框。
装配参数对话框
图:装配体参数对话框

Sequencher使用在此窗口中设置的值来控制其组装序列的方式。 设置选项后,需要单击“确定”按钮以关闭“装配参数”窗口。
组装算法
要选择要使用的组装算法,请在“项目”窗口上单击“组装参数”按钮,然后单击窗口顶部的三个单选按钮之一。您的选择将取决于您的数据。
例如,如果单击“清除数据”单选按钮,重叠群装配会更快,但如果序列的末端包含大量歧义,Sequencher可能会错过某些可能的匹配。为了获得最佳性能,请确保已修剪掉序列中质量较差的数据。
脏数据算法要慢一些,因为Sequencher在序列之间执行的比较严格。您应该注意,自动定序器会创建脏数据,并且Sequencher的算法已针对该问题进行了优化。
大间隙算法使您可以组装预期包含长度超过10个碱基的插入和缺失(间隙)的序列。通常在进化研究和不同生物的序列比较中发现这种缺口。大间隙算法与脏数据算法相似,但速度较慢,但是在执行装配时允许出现较大的间隙。需要大间隙算法的装配的典型示例包括cDNA序列与基因组序列的比较以及相关基因的装配和选择性剪接。
整理脏数据
转到“装配参数”对话框,然后选择“脏数据”单选按钮。有两个滑块可用于更改最小匹配值。要设置必须匹配的碱基比例,请使用“最小匹配百分比”滑块。要设置必须重叠的最小底数,请使用“最小重叠”滑块。
通过将光标放在滑块上,按住鼠标按钮的同时向左或向右拖动滑块来更改设置。移动滑块时,设置的值将自动更新。
例如,如果您尝试组装一个与所选1000个碱基序列完全匹配的17个碱基的序列,并将最小重叠设置为20个碱基(Sequencher的默认最小重叠),则这两个片段将不会组装。您必须首先将最小重叠减少到17个碱基或更低,然后组装片段。
整理干净的数据
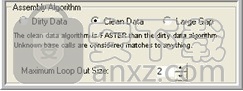
当选择“清理数据”算法时,可以使用一个名为“最大循环输出大小”的附加选项。它具有一个用于调节尺寸值的升降器按钮(下图)。此参数是在潜在重叠中可接受的连续不匹配碱基的最大数量。通过将光标放在滑块上,按住鼠标按钮,并上下拖动滑块来更改滑块设置。可以设置的最大值为6。
要设置必须匹配的碱基比例,请使用“最小匹配百分比”滑块。要设置必须重叠的最小底数,请使用“最小重叠”滑块。
显示最大循环输出大小的Clean Data算法
图:显示最大循环输出大小的清理数据算法
大间隙组装
大间隙算法专门用于具有或可能具有较大插入或间隙的数据。这可能包括来自进化研究,不同生物或将cDNA与基因组DNA进行比较的数据。选择大间隙算法时,有两个滑块可用于更改最小匹配值。要设置必须匹配的碱基比例,请使用“最小匹配百分比”滑块。要设置必须重叠的最小底数,请使用“最小重叠”滑块。
注意:“装配参考”不能与大间隙算法一起使用。
完善装配条件
最低比赛百分比
最小匹配百分比可以与所有三种组装算法一起使用。它用于设置在Sequencher接受序列实际上重叠之前必须在候选序列中匹配的碱基比例。可以通过移动滑块来更改默认值85%。
如果此值不给您任何重叠,则可能需要通过将滑块向左移动以降低百分比来降低“最小匹配百分比”。
如果您有不正确或意外的重叠,则可能需要通过将滑块向右移动(更高的百分比)来提高“最小匹配百分比”。
最小重叠
最小重叠可以与所有三种组装算法一起使用。它用于设置在Sequencher接受序列为实际重叠之前必须重叠的最小碱基数。可以通过移动滑块来更改默认值20。
如果此值不给您任何重叠,则可以通过将滑块向左移动以指示必须重叠的基数较少来减小“最小重叠”。
如果您有不正确或意外的重叠,则可能需要通过将滑块向右移动(必须重叠的碱基数量更多)来增加“最小重叠”。
最大环出尺寸
最大循环输出大小是在潜在重叠中可接受的连续不匹配碱基的最大数量(在这种情况下,间隙也算作不匹配)。通过向上或向下改变电梯来设置不匹配基数。可以手动设置的最大值为6。请记住,此选项只能与Clean Data算法一起使用。
间隙位置的优化
ReAligner是一个可选步骤,可与Clean Data和Dirty Data算法一起使用。它评估重叠群内间隙的分布并优化它们的位置。 ReAligner有助于按共有序列进行编辑,并清楚显示插入和删除的效果。
要使用ReAligner,请选中“装配参数”对话框中的复选框。在DNA分析的某些领域,标准是收集右边的空白。如果需要,请选择“首选3'间隙放置”。
注意:此选项不能与大间隙算法一起使用。
进行组装
自动组装
将项目窗口置于最前面,然后选择要组装的序列片段。单击“自动组装”按钮或进入“重叠群”菜单,选择“组装重叠群”命令,然后从子菜单中选择“自动组装”。 Sequencher比较所有选定的序列,包括反向互补,并组装落入选定组装参数内的最佳匹配。
组装完成后,将显示咨询窗口。 (请参见下图。)单击“关闭”返回到“项目窗口”。
组装完成警报
图:组装完成警报

将选定的项目添加到其他项目–重叠群的增量构建
装配选项“将选定的项目添加到其他”是自动装配的一种特殊情况,当数据集不断增长时非常有用。 Sequencher将所有选中的(新)项目与未选中的(旧)项目进行比较。还将比较新项目。它被设计用来提高处理大量序列的效率,例如DNA库聚类和基因组组装。因此,它将不会再次比较任何未选择的项目。
转到“重叠群”菜单,然后单击“将所选项目添加到其他”,然后单击“组装重叠群”。
组装参考
使用“参考组装”命令,可以将样本组装为单个参考序列,而不管各个片段之间的不一致之处。因为这是一个多对一的比较,而不是正常的多对多比较,所以“参考组装”比标准“自动组装”算法快得多。
要使用此命令,您需要已经指定了参考序列。单击您的序列,然后转到序列菜单,然后选择参考序列。现在,选择要组装的序列片段,包括参考序列。单击“装配为参考”按钮,或从“重叠群”菜单中选择“装配连续体”命令,然后从子菜单中选择“装配为参考”。
互动组装
每当您要完全控制一批序列的组装时,都可以使用“交互式组装”算法。 “交互式组装”窗口为您提供有关任何给定序列和一组候选序列的重叠,不匹配和缺口的详细信息。候选装配由用户定义的装配参数设置驱动。
进行交互式装配
将项目窗口置于最前面,然后选择要组装的项目。单击“项目窗口”顶部的“交互式组装”按钮,或转到“重叠群”菜单,然后从子菜单中选择“组装重叠群”,然后选择“交互式...”。
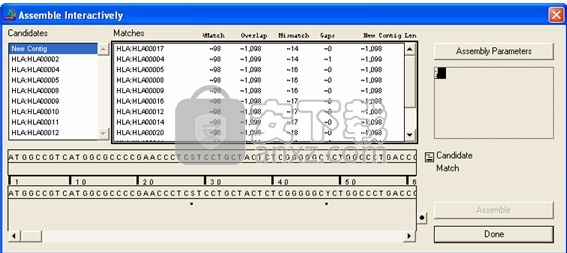
Sequencher将显示“交互式组装”对话框,如下图所示。您选择的序列和重叠群显示在“候选者”列表中。右侧面板在左上角显示一个脸,称为“代理”。它显示有关您要求的比较的信息。中间的“匹配”面板显示可能的匹配。对于面板中的每个匹配项,Sequencher都会列出“%匹配”,“重叠碱基”的近似数,“不匹配碱基”数,“空位”数和“新重叠区长度”。
第一行显示候选序列,而其下方的一行显示匹配序列。在这两行下方是共识序列。共识右侧有一个小按钮,带有黑色圆圈。如果您反复单击此按钮,则可以一次显示第一,第二,第三阅读框或所有三个阅读框。按钮上的标签将更改以反映这些选择中的哪个处于活动状态。
交互式组装对话框
图:“交互式组装”对话框
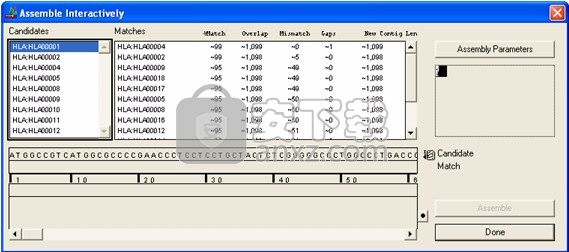
要使Sequencher将任何单个序列或重叠群与“候选”列表中的其他项目进行比较,请在“候选列表”中单击该序列或重叠群的名称。比较完成后,可能的匹配出现在“候选人”列表右侧的“匹配”列表中,如下图所示。
显示候选人及其匹配项的交互式程序集
图:显示候选人及其匹配项的交互式程序集
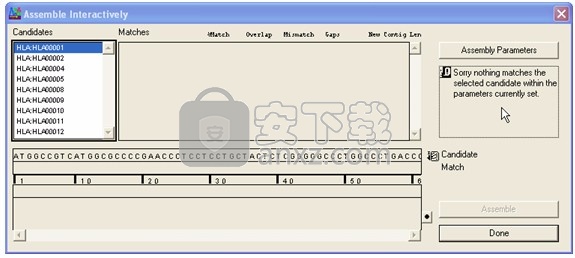
如果Sequencher未能找到匹配项,如下图所示,代理面板将显示一条消息,说明该情况。
找不到匹配项
图:未找到匹配项
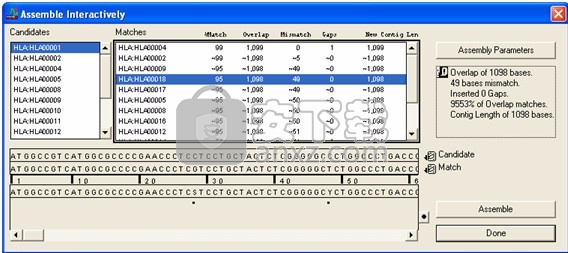
在“匹配”列表中单击可能的匹配时,Sequencher会重新计算缺口的最佳位置。然后,代理会显示有关所选序列及其计算出的重叠的消息。
重叠的开始和基本共识出现在交互式程序集框底部的字段中(下图)。您可以通过向右移动滚动条来进一步探索对齐方式。
交互式装配窗口显示重叠
图:“交互式装配”窗口显示重叠
如果选择创建此装配,请单击“装配”按钮。在“设置名称”对话框窗口中为新重叠群命名,然后单击“确定”。 Sequencher只需很短的时间即可组装(或重新组装)重叠群,而新重叠群的名称现在出现在“候选人”列表的顶部。请注意,在装配序列时,其名称将从“候选人”列表中删除。
在“交互式组装”窗口中显示的新重叠群
图:在“交互式组装”窗口中显示的新重叠群
当您通过单击“完成”按钮关闭“以交互式方式进行装配”对话框时,新的重叠群将出现在“项目窗口”中。
更改交互式装配的参数
如果您怀疑片段应该形成重叠群,但Sequencher无法建议候选匹配,则可以更改组装参数。为此,在使用“交互式装配”时,单击“装配参数”按钮。将出现“装配参数”对话框。将设置更改为满意的设置后,单击“确定”返回到“交互式组装”窗口。
无心加入
Mindlessly Join允许您将序列放在一起,而无需使用常规算法集。您可能希望这样做的情况的例子包括制备质粒或构建人工cDNA序列。
通过在序列周围拖动一个框或按住Shift并单击它们,选择要加入的序列。
注意:它们将按照您选择它们的顺序在新重叠群中列出。
进入“重叠群”菜单,然后选择“组合重叠群”,然后从子菜单中选择“无心加入”。如果启用了“按名称组装”,则“无缝连接”子菜单项将变为“按名称连接”。按名称连接使用句柄将序列适当地分组。
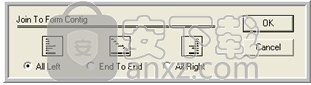
然后,Sequencher会询问您是否要使所有序列在重叠群的左侧(“所有左侧”),重叠群右侧(“所有右侧”)对齐,还是将“端到端”合并。单击适当的按钮。所选项目将被组装,并在项目窗口中被重叠群图标代替。
按名称联接对话框
图:“按名称联接”对话框
按名称组装策略
样品名称经常包含诸如引物,模板,克隆名称或样品来源之类的信息。 Sequencher为您提供了一些工具,用于分隔序列名称的描述性部分并使用它们来管理程序集。这些工具是程序集句柄和名称分隔符。
组装句柄是序列名称中的任何字符集,可提供有关该序列的信息,例如引物或克隆名称。名称定界符将每个组装句柄与下一个分开。序列名称通常具有多个汇编句柄和名称分隔符。
以最简单的形式,名称分隔符可以是诸如破折号或下划线的字符。
使用名称定界符时,它将如下所示:
handle1-handle2-handle3
要么
handle1_handle2_handle3。
但是,名称分隔符也可以是字母,数字和字符的组合。
若要定义名称定界符,Sequencher在“按名称组装”设置对话框中的下拉菜单中提供了一组常用字符。如果转到“程序集参数”并单击“名称设置...”按钮,将看到此菜单。当您的命名约定不使用简单的字符作为“名称分隔符”时,您可以使用称为“正则表达式”的形式化描述符来代替,我们将在本章稍后介绍。
定义装配手柄后,就可以装配项目中的所有序列。仅共享相同装配手柄的序列将被分析以装配到相同重叠群中。例如,在临床应用中,可能会创建多个重叠群,每个重叠群仅代表单个患者的序列数据,前提是每个样品(患者)的名称都具有唯一的装配手柄。
通过名称条件设置装配
使用单个定界符配置程序集句柄
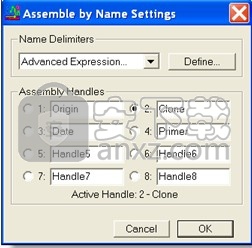
从“项目”窗口开始,单击“装配参数”按钮。在“装配参数”窗口的底部,单击“名称设置...”按钮。下图显示“按名称组装”窗口。
按名称组装设置窗口
图:按名称组装设置窗口
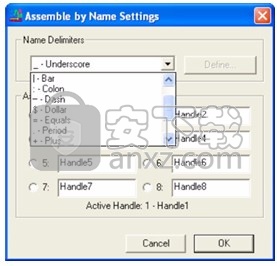
如果“装配句柄”由“名称分隔符”下拉菜单中包含的单个字符分隔,请从列表中选择该字符。否则,请遵循高级表达式的指示。
名称分隔符下拉菜单
图:名称分隔符下拉菜单
什么是正则表达式
正则表达式是一种使用字母,数字和特殊字符描述文本模式的方法。这些表达遵循某些语法规则。
从“项目”窗口开始,单击“装配参数”按钮。在“装配参数”窗口的底部,单击“名称设置...”按钮。出现“按名称组装设置”窗口。
从“名称分隔符”下拉菜单中选择“高级表达式...”。您会注意到“定义...”按钮现已启用。单击定义...按钮。
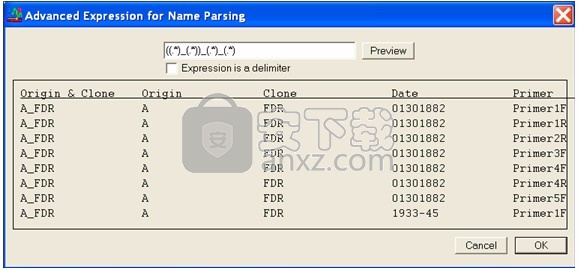
出现“名称解析高级表达式”窗口。窗口顶部是一个文本框,您可以在其中输入正则表达式。
按名称组装使用正则表达式有两种方式。选中“表达式是分隔符”框时,您键入的正则表达式使您可以定义分隔符选项,而不是下拉菜单中可用的选项。
如果未选中“表达式是分隔符”框,则您的正则表达式必须在序列名称中完全定义每个程序集句柄和名称分隔符。除非它描述了序列的全名,否则您编写的正则表达式将不起作用。在文本字段中键入正则表达式时,它应采用以下形式:
(装配手柄1)名称分隔符(装配手柄2)
在下图的示例中,正则表达式用于组合序列名称的前两个部分Origin和Clone,以创建新的Assembly Handle Origin&Clone。预览按钮显示正则表达式的结果。如果满意,请单击“确定”按钮。
名称解析高级表达式对话框窗口
图:“用于名称解析的高级表达式”对话框窗口
设置程序集句柄名称
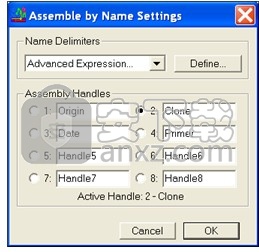
从“项目”窗口开始,单击“装配参数”按钮。 在“装配参数”窗口的底部,单击“名称设置...”按钮。 出现“按名称组装设置”窗口,如下图所示。
您最多可以描述八个装配手柄,在任何时候只有一个处于活动状态。
对于每个Assembly Handle,您可以使用Sequencher自动为您的重叠群自动赋予的默认名称,也可以在文本框中键入描述性标题。 单击要激活的手柄左侧的单选按钮。 Sequencher在此对话框的底部显示活动句柄的编号和名称。 单击确定按钮以退出此窗口并返回到“装配参数”窗口。
“按名称组装”设置窗口
人气软件
-

endnote x9.1中文版下载 107.0 MB
/简体中文 -

Canon IJ Scan Utility(多功能扫描仪管理工具) 61.55 MB
/英文 -

A+客户端(房源管理系统) 49.6 MB
/简体中文 -

第二代居民身份证读卡软件 4.25 MB
/简体中文 -

船讯网船舶动态查询系统 0 MB
/简体中文 -

ZennoPoster(自动化脚本采集/注册/发布工具) 596.65 MB
/英文 -

中兴zte td lte 18.9 MB
/简体中文 -

originpro 2021 527 MB
/英文 -

个人信息管理软件(AllMyNotes Organizer) 5.23 MB
/简体中文 -

ZKTeco居民身份证阅读软件 76.2 MB
/简体中文
 有道云笔记 8.0.70
有道云笔记 8.0.70  Efficient Efficcess Pro(个人信息管理软件) v5.60.555 免费版
Efficient Efficcess Pro(个人信息管理软件) v5.60.555 免费版  originpro8中文 附安装教程
originpro8中文 附安装教程  鸿飞日记本 2009
鸿飞日记本 2009  竞价批量查排名 v2020.7.15 官方版
竞价批量查排名 v2020.7.15 官方版  Scratchboard(信息组织管理软件) v30.0
Scratchboard(信息组织管理软件) v30.0  Fitness Manager(俱乐部管理软件) v9.9.9.0
Fitness Manager(俱乐部管理软件) v9.9.9.0 









